Enabling tools: DNA sequencing

My goal here is to review exciting biological techniques and what discoveries and applications they enable. I write this primarily as an exercise for myself, but hopefully there's something for everyone. This narrative is my personal conception, and suffers from mistakes and omissions that are entirely my own.
Progress in science depends on new techniques, new discoveries, and new ideas, probably in that order. - Sidney Brenner
DNA and the code of life
All life on Earth has settled on DNA as the primary mechanism for encoding and passing on information between generations. DNA is a polymer (long chain) of building blocks, each drawn from a set of four nucleotides written A, C, G, T. These nucleotides come in two pairs of two (A-T and C-G). The chemistry binds two single-stranded DNA sequences that are exact complements of each other into a twisted helix. This design also hints at how DNA is replicated and propagated.

In multicellular organisms, DNA is stored deeply-condensed within the cell nucleus, located near the center of the cell. Because it is a high-fidelity compressed representation of everything that would ever need to be made by any cell in an organism, one can think of an organism's DNA genome as an expansive cookbook.

Each cell has machinery to extract and copy only the cookbook recipes (genes) that are needed for that cell's operation. Continuing the analogy, if DNA is the cookbook, then RNA is a copied recipe extracted from the book. It's produced via the cell's transcription machinery.

After transcription, there is translation machinery that converts the base-4 digital recipe stored in RNA molecules to protein chains. In this way, proteins start off as long chains of amino acids (a total of 20 amino acid building blocks) that fold and assemble into useful products.

Proteins do plenty of interesting things: structural proteins that give cells and tissues their shape, membrane proteins that shuttle molecules across cell membranes, enzymes that catalyze important reactions, and much more.

DNA, RNA, and proteins are the fundamental building blocks of almost everything important in biology. When they become disordered, disease emerges. Any attempt to understand life, biology, and human medicine must proceed from the earliest stages. Thus, we pass to the technologies that enable us to read and write DNA sequences.
Retreading the path to DNA sequencing technologies
Like our ability to fly (which relies on the same physics as birds but looks very different), our ability to sequence DNA derives partially from the natural biology of DNA replication but also looks quite different.
Here, we review three developments in our understanding and control of the DNA replication process:
Polymerase chain reaction (PCR)
Sanger sequencing
Next-gen sequencing (Illumina dye sequencing)
Polymerase chain reaction (PCR), 1983
In the wild, DNA is a small fraction of the total mass of cells. Without a way to amplify at extremely high fidelity naturally-occurring DNA, there would be no way to extract enough DNA for downstream analyses like sequencing. PCR harnesses and accelerates the natural DNA replication process, enabling actually exponential increases of a small amount of DNA within a few hours.

In the natural DNA replication process, double-stranded DNA (dsDNA) gets split into two complementary strands of single-stranded DNA (ssDNA). Each ssDNA strand acts as a template strand for a new copy. After one step, we end up with two identical copies of the initial dsDNA template. Continuing this chain reaction at speed can generate enormous quantities of DNA.
Now, DNA is extraordinarily stable. It has to be, to be able to serve as the key store of genetic information across all species at naturally-occurring temperatures. It does not tend to split apart very easily, unless it is heated to near-boiling temperatures, when the bonds between strands break. You could do a single round of replication by boiling DNA and then adding enzymes for DNA replication... but by the next round of boiling the DNA, you also destroy the enzymes, which are quite expensive.
The key development was the discovery of a special class of DNA polymerase (DNAp) enzymes, extracted from thermophilic bacteria that survive and replicate at the high-temperatures found in hot springs. These DNAp enzymes can survive repeated cycles between room temperature and near-boiling temperatures. By combining these enzymes with the DNA to be amplified and repeatedly cycling the temperature, enormous amounts of DNA can be generated at a more reasonable price point.
Today, a lab is not a lab without PCR machines. PCR is a common step in everything ranging from biology R&D to clinical diagnostics. For example, the gold standard for COVID diagnosis uses PCR to amplify the tiny amounts of viral COVID RNA present in saliva samples to detectable levels.
Sanger sequencing (1980s-present)
Sanger sequencing was the very first commercial DNA sequencing technology. How does it work? How would you build it given the tools available in the 1980s?
Suppose we start with a single short (less than 500 base pairs) sequence of DNA we want to sequence. We apply regular PCR to increase the amount of DNA we have to work with, which requires us to mix a primer (binds to the sequence we want to start DNA replication), DNA polymerase (the enzyme that does DNA replication), and an even mixture of the four nucleotides (let's call them dNTPs).
Sanger sequencing is essentially PCR with a small amount of special fluorescent terminating nucleotides (ddNTPs).
Fluorescent terminating nucleotides (ddNTPs)
If we tag each of the A, C, G, T nucleotides with different-colored fluorescing dyes, then we can use light to help us identify which base pairs were added.
Of course, we don't want to attach dye to every nucleotide in a DNA sequence. We can modify the nucleotides to terminate the elongation process each time they are incorporated. If we add these special nucleotides (ddNTPs) in very small amounts compared to the normal nucleotides, then the reaction will generate random fragments of all lengths as the terminating nucleotides get added. Each of these fragments also fluoresces with the color that corresponds to the most-recently added nucleotide.

Separation by length and detection
How does one separate out these fragments? It means nothing to have a mass of fragments that fluoresce differently without a way of ordering them by length. Diffusion provides exactly what we want.
Gel electrophoresis exploits the fact that longer DNA fragments move more slowly through a dense gel medium. By applying a small voltage across a gel with DNA strands deposited at one end, the small fragments travel further than the big fragments. Doing this for a long time allows the full separation of DNA by their length.
By sending the result of our PCR step through capillaries filled with gel and detecting the fluorescence signal across time, we can get an exact readout of the DNA sequence information as the fragments pass by from smallest to largest.


Sanger sequencing was used in the Human Genome Project to arrive at the very first reference human genome, albeit at enormous cost. Today, it is still used for small amounts of sequencing, like the quality control checks for synthesized DNA fragments.
Era of next-gen sequencing competition (2005-2012)
Next-gen sequencing is an umbrella term referring to everything after Sanger sequencing. After the Human Genome Project was completed in 2003 using slow and expensive Sanger sequencing, government funding for NGS technologies heated up and kicked off an era of intense NGS competition.
Competition between technologies might seem wasteful, but we cannot know which of many early-stage technologies will end up scaling. We simply have to try all of them — and there were quite a few:
454 (sold to Roche in 2007 and shut down in 2013) - pyrosequencing (sequencing by synthesis)
ABI Solid (available since 2006) - sequencing by ligation
Ion Torrent (sold to Life Tech in 2010) - ion-release sequencing by synthesis
Helicos (shut down in 2011) - single-molecule sequencing by synthesis
Pacific Biosciences (available since 2011) - real-time single-molecule long-read sequencing
Oxford Nanopore (available since 2015) - real-time single-molecule long-read sequencing
Solexa (sold to Illumina in 2007) - short-read bulk dye sequencing by synthesis
If one were to classify a “winner” of this era, it would have to be Illumina (acq. Solexa). Their bulk short-read sequencing technology ended up being the powerhouse scalable technology, driving costs from $10M per genome in 2007 to $1K today with a path to $100. Today, you can get your own genome sequenced for $300 [link, no affiliation].

Illumina dye sequencing
Each of these sequencing technologies deserves its own post, but I'm only going to give an overview about how and why Illumina's sequencing technology works. Critically, any discussion of a "$100 genome” implicitly assumes using Illumina or one of its close competitors (eg BGI Genomics).
Sample prep
How does the technology work? We start with a sample of dsDNA, and attach proprietary adapter sequences to both ends of each strand.
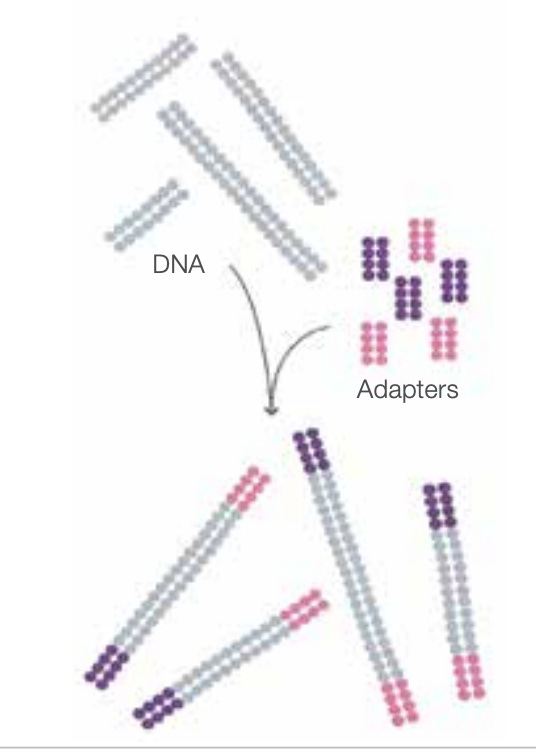
Seeding the flow cell
After denaturing the double-stranded DNA (dsDNA) to ssDNA, flowing the sequences across a prepared flow cell causes them to bind to complementary adapter sequences that were pre-fixed to the flow cell.
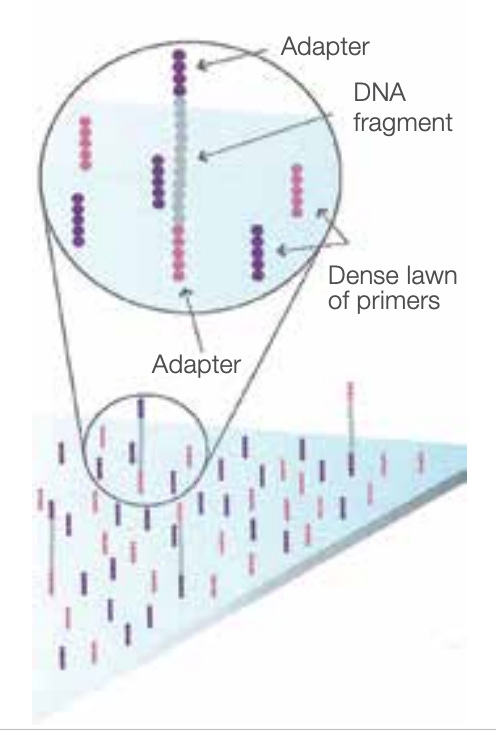
Bridge amplification and cluster generation on the flow cell
We could proceed with sequencing on these attached sequences, but the low density of strands means that the signal-to-noise ratio for each strand is very low. Bridge amplification is a means to generate thousands of copies of the same strand in close proximity to the original strand, generating clusters of copies. Each strand functions as a “vote” during sequencing, and the sheer number of strand copies means that the vote for each base tends to overwhelmingly concentrate on the true base, outweighing any vote for an incorrect base.
Bridge amplification can be thought of as “localized PCR”. Note that there are two unique adapter sequences, one for each end of the DNA. Since each strand is relatively short, each ssDNA can “bridge” so that the non-bound adapter attaches to another. Adding nucleotides and DNA polymerase then generates a complementary strand.

Denaturing this double-stranded bridge leaves us with matching single-strands of DNA attached near each other, but in reverse order. We then repeat this process to generate dense clusters of matching strands.

Note that all this cluster generation relies on nothing but random attachment of sequences to the solid surface of the flow cell and the ability to flow reagents across it. Competitors that relied on mechanical means of controlling strand positioning (micro beads, etched wells, etc) could not compete on density, and therefore had worse scaling.
Sequencing
Finally, we sequence the clusters. We proceed with only one set of the special adapter sequences, so that sequencing proceeds only with the end of the DNA that is farthest from the plate. As in Sanger sequencing, we use fluorescent nucleotides that are also reversible and terminating (RTs). As the nucleotides get incorporated, we image each cluster to get the base that was just sequenced. The fluorescent RTs can be flushed away, the next base added, and the process repeated to get the full sequence.

Preview of the next segment
In the next few segments, I will be covering current and future applications of sequencing technologies, including the following:
Consumer and clinical genotyping, re-sequencing
Genetic testing from 23andme, Color Genomics
Genome-wide association studies and their statistical limitations
Cancer genotyping
Research frontiers
Transcriptomic analysis (RNA sequencing)
Microbiome analysis
Further down the line, I expect to cover the following:
DNA synthesis
Cell therapies
Consumables, reagents, and other complements
Computational drug discovery